Historia
Los antiguos filósofos y médicos griegos y romanos asociaron a la vejez con la demencia. No fue hasta 1901 que el psiquiatra alemán Alois Alzheimer identificó el primer caso de lo que se conoce hoy como enfermedad de Alzheimer, en una mujer de cincuenta años de edad, a quien llamó «Auguste D». El investigador hizo seguimiento de su paciente hasta su muerte en 1906, momento en que por primera vez reportó el caso públicamente. Durante los siguientes cinco años, la literatura médica reportó al menos once casos similares, algunos de ellos utilizando ya el término enfermedad de Alzheimer. La enfermedad fue categorizada por primera vez por Emil Kraepelin después de la supresión de algunos elementos clínicos concomitantes como delirios y alucinaciones, así como características histológicas irrelevantes para la enfermedad como los cambios arterioscleróticos, los cuales figuraban en el informe original sobre Auguste D. En la octava edición de su libro de texto de Psiquiatría, publicado en 1910, incluyó a la enfermedad de Alzheimer, denominada también por Kraepelin demencia presenil, como un subtipo de demencia senil.
EPIMEDIOLOGIA.
La prevalencia es el porcentaje de una población dada con una enfermedad. La edad avanzada es el principal factor de riesgo para sufrir la enfermedad de Alzheimer: mayor frecuencia a mayor edad. En los Estados Unidos, la prevalencia de la EA fue de un 1,6% en el año 2000, tanto en la población general como en la comprendida entre los 65 y 74 años. Se apreció un aumento del 19% en el grupo de los 75 a los 84 años y del 42% en el mayor de 84 años de edad; sin embargo, las tasas de prevalencia en las regiones menos desarrolladas del mundo son inferiores. La Organización Mundial de la Salud estimó que en 2005 el 0,379% de las personas a nivel mundial tenían demencia y que la prevalencia aumentaría a un 0,441% en 2015 y a un 0,556% en 2030. Por otro lado, para el año 2010 la Alzheimer's Disease International ha estimado una prevalencia de demencia del 4,7% a nivel mundial para personas con 60 años o más, representando por cierto cifras al alza respecto a varios estudios publicados con anterioridad (10% superiores a las estimadas para The Lancet en 2005). Otro estudio estimó que en el año 2006, un 0,4% de la población mundial (entre 0,17–0,89%; valor absoluto aproximadamente 26,6 millones o entre 11,4–59,4 millones) se vería afectado por la EA y que la prevalencia triplicaría para el año 2050
Durante la mayor parte del siglo XX, el diagnóstico de la enfermedad de Alzheimer era reservada para las personas entre las edades de 45 y 65 años con síntomas de demencia. La terminología ha cambiado desde 1977 cuando, en una conferencia sobre la EA, se llegó a la conclusión de que las manifestaciones clínicas y patológicas de la demencia presenil y senil eran casi idénticas, aunque los autores también agregaron que ello no descarta la posibilidad que tuviesen causas diferentes. Esto, a la larga, conllevó a que se haga el diagnóstico de la enfermedad de Alzheimer independientemente de la edad. El término demencia senil del tipo Alzheimer fue empleado durante un tiempo para describir al trastorno en aquellos mayores de 65 años, mientras que la enfermedad clásica de Alzheimer se reservaba para los de edades menores. Finalmente, el término enfermedad de Alzheimer fue aprobado oficialmente en la nomenclatura médica para describir a individuos de todas las edades con un patrón de síntomas característica, curso de la enfermedad, y neuropatología comunes.
Etiología
 Las causas de la enfermedad de Alzheimer (EA) no han sido completamente descubiertas. Existen tres principales hipótesis para explicar el fenómeno: el déficit de la acetilcolina, la acumulación de amiloide y/o tau y los trastornos metabólicos.
Las causas de la enfermedad de Alzheimer (EA) no han sido completamente descubiertas. Existen tres principales hipótesis para explicar el fenómeno: el déficit de la acetilcolina, la acumulación de amiloide y/o tau y los trastornos metabólicos.
La más antigua de ellas, y en la que se basan la mayoría de los tratamientos disponibles en el presente, es la hipótesis colinérgica, la cual sugiere que la EA se debe a una reducción en la síntesis del neurotransmisor acetilcolina. Esta hipótesis no ha mantenido apoyo global por razón de que los medicamentos que tratan una deficiencia colinérgica tienen reducida efectividad en la prevención o cura del Alzheimer, aunque se ha propuesto que los efectos de la acetilcolina dan inicio a una acumulación a tan grandes escalas que conlleva a la neuroinflamación generalizada que deja de ser tratable simplemente promoviendo la síntesis del neurotransmisor.[35] [36]
Otra hipótesis propuesta en 1991, se ha relacionado con el acúmulo anómalo de las proteínas beta-amiloide (también llamada amiloide Aβ) y tau en el cerebro de los pacientes con Alzheimer. En una minoría de pacientes, la enfermedad se produce por la aparición de mutaciones en los genes PSEN1, PSEN2 y en el gen de la APP, localizado en el cromosoma 21. En este último caso la enfermedad aparece clásicamente en personas con el síndrome de Down (trisomía en el cromosoma 21), casi universalmente en los 40 años de vida y se transmite de padres a hijos (por lo que existen, habitualmente, antecedentes familiares de enfermedad de Alzheimer en los pacientes que desarrollan la enfermedad en edades precoces). Esa relación con el cromosoma 21, y la tan elevada frecuencia de aparición de la enfermedad en las trisomías de ese cromosoma, hacen que la teoría sea muy evidente.
Patogenia
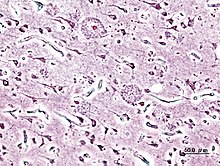
 Imagen histopatológica de placas seniles vista en la corteza cerebral de un paciente con la enfermedad de Alzheimer. Impregnación con plata.La enfermedad de Alzheimer se caracteriza por la pérdida de neuronas y sinapsis en la corteza cerebral y en ciertas regiones subcorticales. Esta pérdida resulta en una atrofia de las regiones afectadas, incluyendo una degeneración en el lóbulo temporal y parietal y partes de la corteza frontal y la circunvolución cingulada.[36]
Imagen histopatológica de placas seniles vista en la corteza cerebral de un paciente con la enfermedad de Alzheimer. Impregnación con plata.La enfermedad de Alzheimer se caracteriza por la pérdida de neuronas y sinapsis en la corteza cerebral y en ciertas regiones subcorticales. Esta pérdida resulta en una atrofia de las regiones afectadas, incluyendo una degeneración en el lóbulo temporal y parietal y partes de la corteza frontal y la circunvolución cingulada.[36]
 Enzimas actuando sobre la proteína precursora de Amiloides (APP) cortándola en fragmentos de beta-amiloide, los cuales son indispensables para la formación de las placas seniles del Alzheimer.La enfermedad de Alzheimer se ha definido como una enfermedad que desdobla proteínas o proteopatía, debido a la acumulación de proteínas Aβ y tau, anormalmente dobladas, en el cerebro.[53] Las placas neuríticas están constituidas por pequeños péptidos de 39–43 aminoácidos de longitud, llamados beta-amiloides (abreviados A-beta o Aβ). El beta-amiloide es un fragmento que proviene de una proteína de mayor tamaño conocida como Proteína Precursora de Amiloide (APP, por sus siglas en inglés). Esta proteína es indispensable para el crecimiento de las neuronas, para su supervivencia y su reparación post-injuria.[54] [55] En la enfermedad de Alzheimer, un proceso aún desconocido es el responsable de que la APP sea dividida en varios fragmentos de menor tamaño por enzimas que catalizan un proceso de proteolisis.[56] Uno de estos fragmentos es la fibra del beta-amiloide, el cual se agrupa y deposita fuera de las neuronas en formaciones microscópicamente densas conocidas como placas seniles.[14] [57]
Enzimas actuando sobre la proteína precursora de Amiloides (APP) cortándola en fragmentos de beta-amiloide, los cuales son indispensables para la formación de las placas seniles del Alzheimer.La enfermedad de Alzheimer se ha definido como una enfermedad que desdobla proteínas o proteopatía, debido a la acumulación de proteínas Aβ y tau, anormalmente dobladas, en el cerebro.[53] Las placas neuríticas están constituidas por pequeños péptidos de 39–43 aminoácidos de longitud, llamados beta-amiloides (abreviados A-beta o Aβ). El beta-amiloide es un fragmento que proviene de una proteína de mayor tamaño conocida como Proteína Precursora de Amiloide (APP, por sus siglas en inglés). Esta proteína es indispensable para el crecimiento de las neuronas, para su supervivencia y su reparación post-injuria.[54] [55] En la enfermedad de Alzheimer, un proceso aún desconocido es el responsable de que la APP sea dividida en varios fragmentos de menor tamaño por enzimas que catalizan un proceso de proteolisis.[56] Uno de estos fragmentos es la fibra del beta-amiloide, el cual se agrupa y deposita fuera de las neuronas en formaciones microscópicamente densas conocidas como placas seniles.[14] [57]
La enfermedad de Alzheimer se considera también una tauopatía, debido a la agregación anormal de la proteína tau. Las neuronas sanas están compuestas por citoesqueleto, una estructura intracelular de soporte, parcialmente hechas de microtúbulos. Estos microtúbulos actúan como rieles que guían los nutrientes y otras moléculas desde el cuerpo hasta los extremos de los axones y viceversa. Cada proteína tau estabiliza los microtúbulos cuando es fosforilado y por esa asociación se le denomina proteína asociada al microtúbulo. En la EA, la tau procede por cambios químicos que resultan en su hiperfosforilación, se une con otras hebras tau creando ovillos de neurofibrillas y, de esta manera, desintegra el sistema de transporte de la neurona.[58]
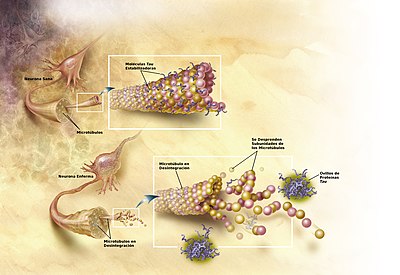 En la enfermedad de Alzheimer, los cambios en la proteína tau producen la desintegración de los microtúbulos en las células cerebrales.No se ha explicado por completo cómo la producción y agregación de los péptidos Aβ juegan un rol en la EA.[59] La fórmula tradicional de la hipótesis amiloide apunta a la acumulación de los péptidos Aβ como el evento principal que conlleva la degeneración neuronal. La acumulación de las fibras amiloides, que parece ser la forma anómala de la proteína responsable de la perturbación de la homeostasis del ion calcio intracelular, induce la muerte celular programada, llamada apoptosis.[60] Se sabe también, que la Aβ se acumula selectivamente en las mitocondrias de las células cerebrales afectadas en el Alzheimer y que es capaz de inhibir ciertas funciones enzimáticas, así como alterar la utilización de la glucosa por las neuronas.[61]
En la enfermedad de Alzheimer, los cambios en la proteína tau producen la desintegración de los microtúbulos en las células cerebrales.No se ha explicado por completo cómo la producción y agregación de los péptidos Aβ juegan un rol en la EA.[59] La fórmula tradicional de la hipótesis amiloide apunta a la acumulación de los péptidos Aβ como el evento principal que conlleva la degeneración neuronal. La acumulación de las fibras amiloides, que parece ser la forma anómala de la proteína responsable de la perturbación de la homeostasis del ion calcio intracelular, induce la muerte celular programada, llamada apoptosis.[60] Se sabe también, que la Aβ se acumula selectivamente en las mitocondrias de las células cerebrales afectadas en el Alzheimer y que es capaz de inhibir ciertas funciones enzimáticas, así como alterar la utilización de la glucosa por las neuronas.[61]
Varios mecanismos inflamatorios y la intervención de las citoquinas pueden también jugar un papel en la patología de la enfermedad de Alzheimer. La inflamación es el marcador general de daño en los tejidos en cualquier enfermedad y puede ser secundario al daño producido por la EA, o bien, la expresión de una respuesta inmunológica.[62]
Los antiguos filósofos y médicos griegos y romanos asociaron a la vejez con la demencia. No fue hasta 1901 que el psiquiatra alemán Alois Alzheimer identificó el primer caso de lo que se conoce hoy como enfermedad de Alzheimer, en una mujer de cincuenta años de edad, a quien llamó «Auguste D». El investigador hizo seguimiento de su paciente hasta su muerte en 1906, momento en que por primera vez reportó el caso públicamente. Durante los siguientes cinco años, la literatura médica reportó al menos once casos similares, algunos de ellos utilizando ya el término enfermedad de Alzheimer. La enfermedad fue categorizada por primera vez por Emil Kraepelin después de la supresión de algunos elementos clínicos concomitantes como delirios y alucinaciones, así como características histológicas irrelevantes para la enfermedad como los cambios arterioscleróticos, los cuales figuraban en el informe original sobre Auguste D. En la octava edición de su libro de texto de Psiquiatría, publicado en 1910, incluyó a la enfermedad de Alzheimer, denominada también por Kraepelin demencia presenil, como un subtipo de demencia senil.
EPIMEDIOLOGIA.
La prevalencia es el porcentaje de una población dada con una enfermedad. La edad avanzada es el principal factor de riesgo para sufrir la enfermedad de Alzheimer: mayor frecuencia a mayor edad. En los Estados Unidos, la prevalencia de la EA fue de un 1,6% en el año 2000, tanto en la población general como en la comprendida entre los 65 y 74 años. Se apreció un aumento del 19% en el grupo de los 75 a los 84 años y del 42% en el mayor de 84 años de edad; sin embargo, las tasas de prevalencia en las regiones menos desarrolladas del mundo son inferiores. La Organización Mundial de la Salud estimó que en 2005 el 0,379% de las personas a nivel mundial tenían demencia y que la prevalencia aumentaría a un 0,441% en 2015 y a un 0,556% en 2030. Por otro lado, para el año 2010 la Alzheimer's Disease International ha estimado una prevalencia de demencia del 4,7% a nivel mundial para personas con 60 años o más, representando por cierto cifras al alza respecto a varios estudios publicados con anterioridad (10% superiores a las estimadas para The Lancet en 2005). Otro estudio estimó que en el año 2006, un 0,4% de la población mundial (entre 0,17–0,89%; valor absoluto aproximadamente 26,6 millones o entre 11,4–59,4 millones) se vería afectado por la EA y que la prevalencia triplicaría para el año 2050
Durante la mayor parte del siglo XX, el diagnóstico de la enfermedad de Alzheimer era reservada para las personas entre las edades de 45 y 65 años con síntomas de demencia. La terminología ha cambiado desde 1977 cuando, en una conferencia sobre la EA, se llegó a la conclusión de que las manifestaciones clínicas y patológicas de la demencia presenil y senil eran casi idénticas, aunque los autores también agregaron que ello no descarta la posibilidad que tuviesen causas diferentes. Esto, a la larga, conllevó a que se haga el diagnóstico de la enfermedad de Alzheimer independientemente de la edad. El término demencia senil del tipo Alzheimer fue empleado durante un tiempo para describir al trastorno en aquellos mayores de 65 años, mientras que la enfermedad clásica de Alzheimer se reservaba para los de edades menores. Finalmente, el término enfermedad de Alzheimer fue aprobado oficialmente en la nomenclatura médica para describir a individuos de todas las edades con un patrón de síntomas característica, curso de la enfermedad, y neuropatología comunes.
Etiología

La más antigua de ellas, y en la que se basan la mayoría de los tratamientos disponibles en el presente, es la hipótesis colinérgica, la cual sugiere que la EA se debe a una reducción en la síntesis del neurotransmisor acetilcolina. Esta hipótesis no ha mantenido apoyo global por razón de que los medicamentos que tratan una deficiencia colinérgica tienen reducida efectividad en la prevención o cura del Alzheimer, aunque se ha propuesto que los efectos de la acetilcolina dan inicio a una acumulación a tan grandes escalas que conlleva a la neuroinflamación generalizada que deja de ser tratable simplemente promoviendo la síntesis del neurotransmisor.[35] [36]
Otra hipótesis propuesta en 1991, se ha relacionado con el acúmulo anómalo de las proteínas beta-amiloide (también llamada amiloide Aβ) y tau en el cerebro de los pacientes con Alzheimer. En una minoría de pacientes, la enfermedad se produce por la aparición de mutaciones en los genes PSEN1, PSEN2 y en el gen de la APP, localizado en el cromosoma 21. En este último caso la enfermedad aparece clásicamente en personas con el síndrome de Down (trisomía en el cromosoma 21), casi universalmente en los 40 años de vida y se transmite de padres a hijos (por lo que existen, habitualmente, antecedentes familiares de enfermedad de Alzheimer en los pacientes que desarrollan la enfermedad en edades precoces). Esa relación con el cromosoma 21, y la tan elevada frecuencia de aparición de la enfermedad en las trisomías de ese cromosoma, hacen que la teoría sea muy evidente.
Patogenia
_presenile_onset.jpg)
[editar] Neuropatología
Las placas son depósitos densos, insolubles, de la proteína beta-amiloide y de material celular que se localizan fuera y alrededor de las neuronas. Estas continúan creciendo hasta formar fibras entretejidas dentro de la célula nerviosa, los llamados ovillos. Es probable que muchos individuos, en su vejez, desarrollen estas placas y ovillos como parte del proceso normal de envejecimiento, sin embargo, los pacientes con Alzheimer tienen un mayor número en lugares específicos del cerebro como el lóbulo temporal.[52][editar] Bioquímica

La enfermedad de Alzheimer se considera también una tauopatía, debido a la agregación anormal de la proteína tau. Las neuronas sanas están compuestas por citoesqueleto, una estructura intracelular de soporte, parcialmente hechas de microtúbulos. Estos microtúbulos actúan como rieles que guían los nutrientes y otras moléculas desde el cuerpo hasta los extremos de los axones y viceversa. Cada proteína tau estabiliza los microtúbulos cuando es fosforilado y por esa asociación se le denomina proteína asociada al microtúbulo. En la EA, la tau procede por cambios químicos que resultan en su hiperfosforilación, se une con otras hebras tau creando ovillos de neurofibrillas y, de esta manera, desintegra el sistema de transporte de la neurona.[58]
[editar] Patología

Varios mecanismos inflamatorios y la intervención de las citoquinas pueden también jugar un papel en la patología de la enfermedad de Alzheimer. La inflamación es el marcador general de daño en los tejidos en cualquier enfermedad y puede ser secundario al daño producido por la EA, o bien, la expresión de una respuesta inmunológica.[62]
No hay comentarios:
Publicar un comentario